


![]() L'existence des maladies rares pose un problème de société très souvent mal appréhendé par le public et les autorités. 6 à 8 % de la population, soit environ 4 millions de personnes en France, sont atteintes d'une maladie rare. Compte tenu de leur rareté, et donc du manque d'expérience et d'information des praticiens, leur diagnostic n'est pas toujours évident.
L'existence des maladies rares pose un problème de société très souvent mal appréhendé par le public et les autorités. 6 à 8 % de la population, soit environ 4 millions de personnes en France, sont atteintes d'une maladie rare. Compte tenu de leur rareté, et donc du manque d'expérience et d'information des praticiens, leur diagnostic n'est pas toujours évident.
![]() L'histiocytose langerhansienne, bien que répertoriée depuis longtemps sous des noms variés, reste encore à ce jour, peu ou mal connue. Il est d'autant plus important de la faire connaître des praticiens non-spécialistes que la précocité de son diagnostic est un facteur essentiel au succès de son traitement.
L'histiocytose langerhansienne, bien que répertoriée depuis longtemps sous des noms variés, reste encore à ce jour, peu ou mal connue. Il est d'autant plus important de la faire connaître des praticiens non-spécialistes que la précocité de son diagnostic est un facteur essentiel au succès de son traitement.
![]() Le Groupe d'Etude des Histiocytoses (G.E.H.), créé en 1995, regroupe des médecins et des biologistes français spécialistes de cette maladie rare.
Le Groupe d'Etude des Histiocytoses (G.E.H.), créé en 1995, regroupe des médecins et des biologistes français spécialistes de cette maladie rare.
![]() L'un d'entre eux, le Docteur Jean Donadieu, est pédiatre au service d'hémato-oncologie du Professeur Leverger à l'hôpital Trousseau. Membre fondateur du G.E.H. dont il fut Président de 1995 à 2001, il assure aujourd'hui, au sein du bureau de cette association de chercheurs français, la coordination du registre des patients. Il participe également aux activités de l'association internationale Histiocyte Society dont il est membre du bureau.
L'un d'entre eux, le Docteur Jean Donadieu, est pédiatre au service d'hémato-oncologie du Professeur Leverger à l'hôpital Trousseau. Membre fondateur du G.E.H. dont il fut Président de 1995 à 2001, il assure aujourd'hui, au sein du bureau de cette association de chercheurs français, la coordination du registre des patients. Il participe également aux activités de l'association internationale Histiocyte Society dont il est membre du bureau.
![]() Il a bien voulu répondre aux questions que l'Association Histiocytose France (A.H.F.) lui a posées sur cette maladie rare.
Il a bien voulu répondre aux questions que l'Association Histiocytose France (A.H.F.) lui a posées sur cette maladie rare.
Histiocytose désigne une maladie correspondant à une prolifération d'histiocytes, cellules ayant la forme de macrophages (catégorie de cellules du système immunitaire), quel que soit l'organe atteint.
Langerhansienne qualifie le type de cellules d'aspect macrophagique impliqué dans la maladie. Dans le cas de l'histiocytose langerhansienne (H.L.), les cellules pathologiques sont des cellules de Langerhans localisées, à l'état normal, au niveau de la peau, mais qui dans la situation de la maladie H.L., se localisent parfois dans d'autres organes et sont responsables d'une série de désordres.
Ce nom a lui-même toute une longue histoire. D'autres noms ont été utilisés et le sont parfois encore :
Granulome à éosinophiles désigne une tumeur des os dont l'aspect histologique est associé à des cellules de Langerhans et à de nombreuses cellules réactionnelles, particulièrement des polynucléaires éosinophiles.
La maladie de Hand-Shüller-Christian, du nom des trois médecins qui l'ont décrite, est une forme clinique que l'on rencontre chez l'enfant après un an. Cette forme associe des tumeurs osseuses (granulome à éosinophiles), localisées au niveau du crâne, à un diabète insipide et une exophtalmie (yeux semblant sortir du crâne).
La forme de Letterer-Siwe est une forme du nouveau-né décrite par deux médecins dans les années 20. Cette forme comporte une atteinte hématopoïétique de la moelle osseuse, des os, du foie, de la rate. C'est une forme très sévère qui met en jeu la vie de l'enfant en l'absence de traitement.
La forme de Hashimoto-Pritzker correspond à une forme cutanée pure du tout petit enfant. Elle est réputée être régressive spontanément.
Ces entités diverses ont été rassemblées en 1953. Il s'avère en effet que celles-ci, très différentes dans la présentation clinique, présentent toutes un aspect identique sur le plan tissulaire, c'est-à-dire une accumulation d'histiocytes. Dans la mesure où le type d'histiocytes impliqués n'était pas déterminé, ces entités ont été désignées par le terme Histiocytose X (histiocytes d'origine inconnue).
En 1973, des chercheurs français, Mme Basset et M. Nezelof ont rattaché cette maladie à une cellule de la peau, la cellule de Langerhans, qui porte le nom d'un médecin allemand, Paul Langerhans ; il l'avait décrite dans les années 1870. C'est, à l'état normal, une cellule localisée dans la peau qu'il avait rattachée initialement au système nerveux, en raison de sa forme : en effet, elle présente normalement des ramifications - comme de multiples bras - (morphologie dendritique, du grec dendron, arbre).
On a compris que cette cellule de Langerhans était vraiment le cœur du problème dans différentes entités. D'où le nom d'Histiocytose langerhansienne.
La localisation la plus fréquente est osseuse. Dans les os, le symptôme qui conduit à découvrir cette maladie est la douleur, ou parfois des fractures dites pathologiques parce que survenant sans effort particulier. Dans ce cas, la maladie au niveau des os est caractérisée par une image radiologique très particulière qu'on appelle une lacune osseuse et qui ressemble à un trou dans l'os. C'est la première localisation par la fréquence.
La localisation qui vient au deuxième rang, par la fréquence, se situe au niveau de la peau. L'atteinte cutanée peut présenter plusieurs aspects. Le plus fréquent est constitué par des croûtes de lait sur le cuir chevelu des nouveau-nés. Elle peut aussi prendre la forme d'une éruption parfois dite pétéchiale : de petits points violacés sont répandus sur la peau. On peut encore trouver un aspect de pseudo varicelle (bien entendu non liée au virus de cette maladie).
Dans les autres organes, la maladie s'exprime de façon tout à fait différente.
Par exemple, au niveau du poumon, elle peut se manifester par une gêne respiratoire, et en radiographie par ce qu'on appelle un infiltrat (lésion pulmonaire très diffuse) ou par des bulles.
Si elle atteint le foie, elle entraîne des dysfonctionnements de cet organe, et son grossissement décelable à la palpation.
Elle peut se manifester au niveau de la moelle des os.
Elle peut se localiser au niveau du système endocrinien, avec en particulier une entité appelée diabète insipide. Il s'agit d'un manque de contrôle de la perte urinaire d'eau avec compensation par des boissons abondantes, ou, en jargon médical une polyurie-polydipsie, n'ayant rien à voir avec le diabète sucré dû à un manque d'insuline.
Les localisations neurologiques sont exceptionnelles. Elles présentent soit un aspect de tumeurs cérébrales (de "boules" dans le cerveau) ; soit des aspects considérés comme dégénératifs, avec en particulier un syndrome cérébelleux entraînant un problème de coordination de la statique, de la marche (comme dans un état ébrieux).
Globalement, la plupart des localisations sont uniques (celles de l'os et de la peau en particulier).
Mais pour chaque personne impliquée, cela peut être différent. Il y a quelques formes où la maladie atteint plusieurs organes. On les dit multisystémiques. Ces formes posent avec le plus d'acuité le problème du traitement, car la maladie peut alors être dangereuse pour la santé de l'enfant, et parfois même pour sa vie.
Cette maladie peut atteindre les adultes. Des enfants ont atteint l'âge adulte avec cette maladie et continuent d'avoir des localisations. C'est un tout petit nombre, mais ça existe. Il y a aussi les adultes qui déclenchent une maladie en tout point similaire à celle de l'enfant. Il y a enfin une forme de l'adulte qui est tout à fait remarquable parce que très liée au tabac. Le tabac a beaucoup d'inconvénients ; mais en plus, certaines personnes ayant probablement une susceptibilité à le faire, présentent une atteinte pulmonaire caractérisée par une gêne respiratoire, par des accès de toux, par ce qu'on appelle des pneumothorax (présence d'air dans la plèvre) et des douleurs.
L'origine virale a été suspectée depuis le début de la description de la maladie. Un des arguments en faveur de cette hypothèse tient à la fonction immunitaire des cellules de Langerhans qui constituent un des premiers maillons de la défense anti-infectieuse (ce sont des cellules dites présentatrices d'antigène). Mais malgré les très nombreux travaux effectués, aucune infection virale n'a été mise en évidence de façon récurrente chez ces patients.
La difficulté suivante est de décider d'un traitement particulier. Je pense surtout à la chimiothérapie (types de traitements usuellement utilisés contre le cancer). Quand on a recours à ce type de médication, ces enfants ou ces adultes vont consulter des services de cancérologie. Mais ce n'est pas pour autant que cette maladie est un cancer. Ces services sont amenés à voir et à traiter les formes les plus sévères, qui mettent parfois en jeu la vie des enfants, cette maladie étant, avant tout, une maladie de l'âge pédiatrique.
Pour entrer dans le détail, c'est une maladie très hétérogène, qui peut atteindre de très nombreux organes ; qui a une évolution très variable d'une personne à l'autre, d'un enfant à l'autre. La première considération importante, c'est que le traitement doit être adapté à la sévérité de la maladie et à son profil évolutif. Cela explique que souvent la première étape du traitement est une étape d'observation et d'attente. Ceci est peut-être un peu difficile à accepter quand on est patient, mais cette étape est indispensable parce qu'il y a des formes qui peuvent auto-involuer ou s'arrêter toutes seules.
Quand un traitement s'avère nécessaire, il doit être proportionnel à la gravité de la maladie. Il faut donc vraiment réfléchir au cas par cas ; ou du moins essayer de faire des catégories de patients à traiter de façon un peu homogène, tout en tenant compte de la gravité potentielle de chaque cas. Le schéma thérapeutique doit associer différentes approches :
Pour la peau, on utilise une chimiothérapie spécifique qui consiste en l'application de caryolysine. C'est un traitement qui rend de grands services pour les lésions cutanées.
Dans les formes vraiment agressives de la maladie, on envisage des associations de médicaments. Parmi ces médicaments, on retrouve de façon presque obligatoire deux médicaments, la cortisone ou les dérivés de la cortisone ; et la vinblastine - qui est un alcaloïde -, médicament dérivé d'une plante, la pervenche de Madagascar. Cette vinblastine a été synthétisée et expérimentée pour la première fois dans les années 60. On a donc assez de recul pour savoir que ce médicament est en général parfaitement toléré, qu'il n'est pas dangereux sur le long terme, qu'il a quelques effets secondaires à court terme qu'on peut maîtriser. Il est donc possible de l'utiliser assez facilement chez les enfants et pendant tout le temps qu'il faut pour traiter et venir à bout de cette maladie.
Des traitements de deuxième ou troisième ligne sont à mettre en œuvre si l'on voit que la deuxième approche n'est pas suffisante. On entre ici dans des détails que je ne développerai pas.
En résumé, le traitement, si la décision d'abstention thérapeutique n'est pas retenue, sera soit un traitement local par caryolysine, soit un traitement par vinblastine et corticoïde de première intention ; ensuite, il faudra voir en fonction de la réponse et de l'évolution sous ces traitements.
Une fois ce travail préliminaire effectué, il est possible d'orienter le traitement de façon la plus rationnelle possible compte tenu de ce que l'on sait aujourd'hui. Peut-être y aura-t-il un jour des techniques pour orienter rapidement le traitement. Mais aujourd'hui, on se sert des éléments recueillis par l'examen clinique (l'observation de l'enfant) et par des examens complémentaires : scanners, éventuellement IRM, et différentes prises de sang. En fonction de ces éléments, on stratifie, on organise le traitement selon différents groupes de patients ; on décrit deux groupes ou trois groupes de patients.
Pour certains, on décide de ne rien faire, et on observe. Il s'agit en général de formes osseuses isolées. Il y a des formes intermédiaires où l'on voit que la maladie est quand même assez active, en particulier dans les formes osseuses où plusieurs os sont atteints ; ou des formes osseuses qui menacent éventuellement l'œil ou la colonne vertébrale. Dans ces cas-là, on met en route un traitement, mais d'intensité modérée. Puis, il y a les formes les plus sévères d'emblée qui sont les formes du tout petit enfant où existe une atteinte du poumon, du système sanguin. Ces atteintes sont potentiellement graves pour l'enfant. Pour ces cas-là, on a intérêt à mettre en route sans délai un traitement assez vigoureux.
Elle avance mais très lentement pour plusieurs raisons liées à la maladie elle-même. Usuellement plusieurs moyens existent pour faire progresser les connaissances sur les maladies. Un des moyens est le modèle animal de la maladie qui correspond exactement à la maladie humaine. Mais cela n'est pas réalisable dans le cas de l'histiocytose. L'autre moyen est la culture des tissus porteurs de la maladie ; mais en raison de contraintes biologiques, nous n'arrivons pas à reproduire cette maladie en éprouvette. On est donc obligé d'avancer sur des prélèvements faits lors du diagnostic. Cela nous oblige à travailler sur de toutes petites quantités de tissus. Il faut avec cela essayer de débobiner tous les fils de cette maladie. Ce n'est pas quelque chose de très simple. Et ce l'est d'autant moins que cette maladie étant rare, ces prélèvements sont dispersés. Il y a donc toute une série de problèmes d'ordre logistique à résoudre avant de commencer à réfléchir sur le fonctionnement de cette maladie.
En 2005, les travaux de recherche fondamentale se sont fédérés, avec le réseau de recherche clinique pour un projet dénommé EPILCH2005. Il associe le réseau clinique (c’est-à-dire le registre des patients), et 3 équipes de recherche, une d’épidémiologie (registre des leucémies et lymphomes de l’enfant), une de virologie (Unité Inserm, Madame Joab, Hôpital Saint-Louis) et une d’immunologie et de génétique impliquée dans des travaux sur les cellules dendritiques (Unité Avenir Inserm, Laboratoire d’Anapath, Docteur Geissmann, Hôpital Necker).
Ce projet de 3 ans est financé par le GIS maladies rares et des fonds de l’Agence Nationale de la Recherche. Il vise à connaître les déterminants de l’apparition de l’histiocytose.
Dans les faits, le système immunitaire suppose une coopération de ces différents acteurs. En particulier, la défense immunitaire spécifique (assurée par les lymphocytes) suppose une mise en action d'une partie du système immunitaire non spécifique. Celle-ci est constituée par les cellules dites présentatrices d'antigène ; ce sont très précisément les cellules dites dendritiques, dont font partie les cellules de Langerhans. L'importance de ces cellules présentatrices d'antigène a été longtemps ignorée car elles sont difficiles à étudier. Mais il s'avère qu'elles ont une part importante dans plusieurs situations, par exemple dans la défense contre les infections par certains microbes comme la tuberculose, mais aussi dans le cancer. Il se trouve que l'histiocytose langerhansienne est pratiquement la seule maladie humaine de ce système cellulaire. Arriver à comprendre cette maladie représente un outil fort utile pour comprendre la biologie de ces cellules et les moyens de les réguler, dans le cas de l'histiocytose langerhansienne, mais aussi, pourquoi pas, dans d'autres circonstances bien plus fréquentes comme la défense immunitaire anticancéreuse par exemple.
L'impact potentiel de la compréhension de ce type de maladies apparaît donc assez large.
En 1868, Paul Langerhans décrit des cellules de la peau,
pourvues de ramifications ou dendrites.
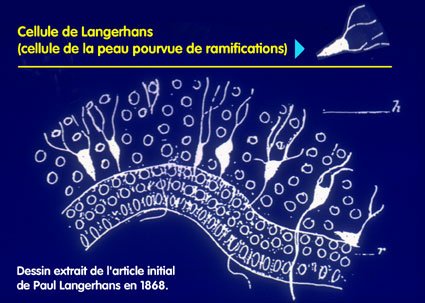
Agé de 23 ans au moment de la publication de cet article,
il pensait que ces cellules étaient des cellules nerveuses,
et ce n'est que 50 ans après que ces cellules
ont été rattachées au système immunitaire.

Paul Langerhans